Typical morning: I eat breakfast with my older sister, some days I enjoy my breakfast, other days I don’t have much appetite and I eat breakfast gradually throughout the morning. Mum and dad have to make sure I have eaten because I’m only able to fast for 10 hours over night to make sure my blood sugar levels don’t drop too low. I love listening to my tonies (audio stories) with my sister at breakfast time.
Typical afternoon: After my midday nap, I usually play at home or out and about. I have a special yogurt to eat with lunch or as a snack which gives me extra calories and nutrients to help me grow.
Typical evening:After dinner, my sister and I watch my favourite tv show Pocoyo. This helps keep me still whilst I have my daily injection. My injection doesn’t bother me, I hardly notice it. Pocoyo helps to distract me. My family are really proud of how brave I am. I have some extra vitamins and formula before bed to make sure I’m getting everything I need.
My favourite thing to do is:I love shoes, I’m always trying on different shoes I can find at home. I also love climbing and going on my bike. I love trucks and cars and one of my favourite things to do is go outside and look at the vehicles pass by.
My least favourite thing is: I don’t like brushing my teeth! This is way worse than my daily injection!
The best thing about having GHD is: I’m lucky that my growth condition can be treated and I am under a really supportive medical team to help me grow and develop. My growth condition has given me resilience and I will grow up knowing I can face any challenge in life.
The most challenging thing about having GHD is: I’ve got lots of medical appointments and sometimes I get anxious, but I’m getting more used to doctors and nurses.
One piece of advice I would give to someone who has this growth condition and/or is a parent carer of a child with this growth condition is: Don’t forget to enjoy your child in the present because you’re constantly worried about what their future will look like.
One thing I want to share with others about life with a growth condition is: Comments about height can have a profound impact on individuals with a growth condition, especially when those comments are insensitive or repetitive.
Fighting for A Growth Hormone Deficiency Diagnosis
This is Jake’s story as told by his mum, Faye. It was at 8 months old, when Jake started nursery, that Faye believed that he could have a growth condition. It was a fight for Faye and her husband to be taken seriously, but through perseverance Jake had his first consultation at two years old.
Our story began in April 2008 when our baby boy, Jake was born. He was 2 weeks overdue and weighed only 5 pounds and 13 ounces. We were a little surprised he was so tiny given how late he was and we had to rush round and get tiny baby clothes rather than the regular ones. Aside from the fact Jake had mild jaundice; we were not too worried as the paediatrician did not think it was a problem. He actually stated that it was good as Jake was unlikely to be ‘obese’.
Jake had quite bad colic which lasted to 12 weeks. Nothing seemed to work for him. We now believe that Jake had silent reflux as this is quite common with small babies, but this was never discussed with us. He also struggled to eat when the time came to wean him. He was simply unable to tolerate lumps. We had to keep pureeing his food. Again, we believe now that this was also due to his small size.
Jake started nursery when he was 8 months old. At first we didn’t realise how little Jake was, but when the other babies the same age as Jake began to walk and Jake didn’t, we had an inkling something was not right. Although Jake was very bright, he was always slightly behind his friends in the physical aspect — the last one to hold his head up, the last one to sit up, and he finally began walking at 16 months. This was the time that Jake was due to move into the ‘toddler’ room at nursery from the ‘baby’ room. The staff kept him back in the baby room for a little longer than normal as he was so small compared to the other children. This is when we raised the issue with one of Jake’s health visitors.
The first health visitor we went to was ok — not overly eager to measure him, but when she did he fell more than two standard deviations below his projected percentile range, she agreed he should be referred to a paediatrician. However, a referral would not be made until he had two measurements, so the health visitor advised that we should return a month later for Jake’s next measurement. We had also booked an appointment with the doctor to discuss Jakes eczema. I raised the issue there and was simply told “don’t worry, Tom Cruise is short but he has done OK”. This is where our trouble began….
I took Jake back a month later but the health visitor refused to measure him. She said that there was nothing wrong with Jake and that ‘unfortunately he has taken after me’ — I am only 5 ft tall. When I stated that I was simply there to have Jake measured as instructed by the previous health visitor, she told me that not enough time had elapsed anyway and there needed to be a gap of at least 3 months between measurements. To cut a long story short, we went back several times and several times she refused to measure him saying the same and that there is nothing wrong with Jake. However, one day my husband took Jake along and the health visitor had a trainee with her and she told my husband that if we didn’t believe her, we should look on the Child Growth Foundation website where it will tell us that there is nothing wrong with Jake. This is the first time she ever mentioned the Child Growth Foundation and we believe that she only mentioned it as she had a trainee with her. But this is where we got the first glimmer of hope.
I am a naturally curious person and I will research something as far as I can. So rather than check the website, I emailed the Child Growth Foundation with Jake’s details. I soon received a reply from Jenny Child stating that given Jake’s details, he should be referred to an endocrinologist. I firmly believe that if we had not got that reply from Jenny, we would not be where we are today (which you will hear about in a bit!). I had done some research on child growth disorders and as Jake had no other apparent symptoms; I believed that he was an SGA baby (Small for Gestational Age). I looked into all the different hospitals in the area and what each endocrinology department specialised in. Here I came across the Royal London. Rather than take this information to Jake’s doctor, I called the hospital personally. I have to say, the secretary there was fantastic. She took all Jake’s details and passed them onto one of the consultants. She called me back to say that Jake definitely fitted the criteria for a referral and she gave me the consultant’s name and fax number.
Both my husband and I attended an appointment with one of our GP’s armed with all the research we had. When we asked for a referral to an endocrinologist, the GP laughed at us and asked us what one was, implying that we had no idea what we were talking about. So I gave her all my research and she just stated that the Royal London was out of our area so she couldn’t refer Jake there. She said that she could refer Jake to a paediatrician but that they wouldn’t do anything as children under the age of 13 are not treated with growth hormones. So we contacted the Royal London again and they confirmed that they are treating children from 18 months upwards and that they do have referrals from our area. They advised that we should make a complaint which was exactly what we did and we got our referral for Jake.
So we ended up seeing the consultant when Jake was two years old. The consultant agreed that Jake was far smaller than he should be but that he may have some catch up growth but they would monitor this. If he had no catch up growth by the time he was four years old, growth hormone treatment could be considered.
So in February 2012, just before Jake’s fourth birthday, we took him to the Royal London for a glucagon stimulation test. This is where glucagon is injected and then blood is taken every half an hour and this is designed to measure how much growth hormone the pituitary gland is capable of making. We were dreading this, but Jake handled it really well and it was just the hunger and the boredom for all of us that proved to be a pain!
Four months later, we took Jake back to the Royal London for the results of his Growth Hormone Stimulation Test. My husband and I (well, more me…) got hyped up ready for a battle. We were convinced that Jake was going to be borderline deficient and therefore wouldn’t qualify for treatment. The doctor was fantastic and told us straight away that the test showed his pituitary gland produces ‘subnormal’ amounts of growth hormone. And because he was also an SGA baby he qualified for treatment on two counts, and he only needed to qualify on one count to receive treatment.
Then came the question and answer session with the doctor — does the treatment carry any risks, what are the side effects, does it hurt etc. We were very pleased with the answers we got. There are very few, if any, risks or side effects, and it is like a little pinch. Lovely Lee, our Endo nurse, gave us four DVD’s to take home and watch to decide what injection device we would like to use for Jake. Talk about too much information! Right away we were keen on the Easypod. I liked the fact it was a gadget that made everything really simply. You just programme in, insert a needle, inject, and take off the needle. We thought it would make it really easy for Jake’s Nana’s.
I had thought that GHD only affected height but it affects so much more — bone and muscle strength, cardiovascular system, delayed puberty, increased fat, increased cholesterol. This made a lot of sense. Jake had always struggled with walking and constantly complained of his legs hurting. We initially thought that he just didn’t want to walk anywhere. But Jake was visibly distressed and I now know that his legs were simply not strong enough. He also started getting eye infections that would not go away despite lengthy courses of antibiotics.
Having learnt all that, it makes me really angry that we had to fight the health visitor and the GP all that time ago. We were treated like neurotic parents and it’s only because I did my research that we are where we are today.
After my initial euphoria, I was left feeling quite emotional and sad for the fact that Jake would have to have injections every night for years and that if he didn’t, he could become quite poorly.
A month later, we began Jake’s injections. The first few days were extremely traumatic. Jake hated the injections and was physically frightened of me when I had the Easypod in my hands. It really is horrendous having to hurt your child — even though I knew it was the right thing to do; it went all against my instincts and was so hard. But I had established a good network through the Child Growth Foundation and we were given lots of tips to make things easier for us including distraction techniques. I opted to hide some ‘treasure’ (sweets) so that after his injection, he had to go and find them which took his mind off the injection. This did work well. Even though Jake would cry the whole way through the injection, this would be short lived and he certainly wasn’t traumatised.
Jake has now been on injections for three and a half years. He has totally transformed. He was a tired boy lacking in energy and not remotely boisterous which we had put down to just his character. Now he is lively and energetic — he loves riding his scooter, has learnt to ride his bike and even wants to go for runs. One of his most favourite things is playing fighting games with his dad.
Whilst he is still not tall, he is no longer the smallest in his peer group. He has gone from the 0.4th percentile to the 20th percentile. He rarely gets ill now and has had no more eye infections since he started on the injections.
We have so much to thank the Child Growth Foundation for. Without their support, I am convinced that Jake would not be the child he is today. They supported us through the referral stage and then supported us through the initial stages of treatment. They are always there for us and we will be forever grateful for the amazing work that they do.
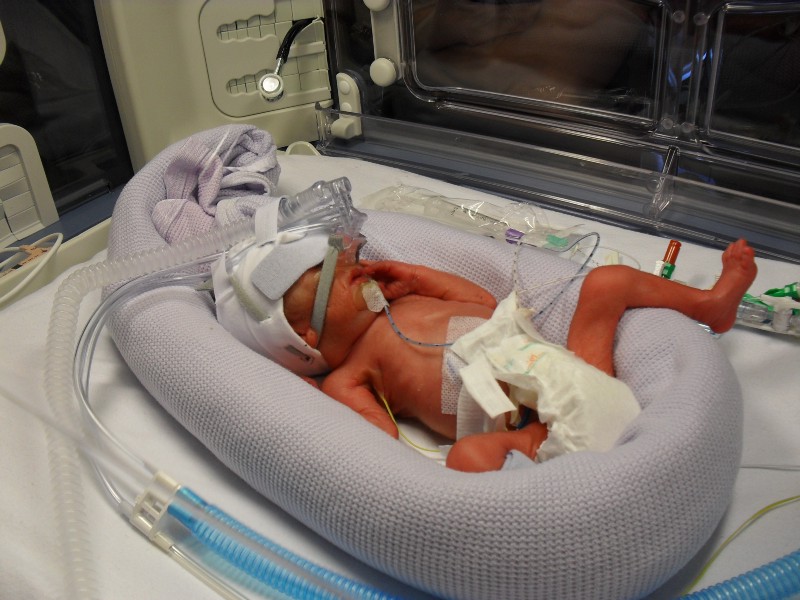
On the 30th March 2011, 11 weeks before his due date and weighing 820grams (1lb 13ozs), Michael made a dramatic entry into the world.
Two days earlier during a routine scan at Solihull hospital, the team discovered that Michael was not growing and probably hadn’t done so for two to three weeks due to an absent end-diastolic flow. I was immediately transferred to Heartlands hospital in Birmingham where they promptly diagnosed that I was suffering from Pre-eclampsia.
Following a forty-five minute emergency caesarean section operation performed by an eighteen person medical team led by Mr Mike Wyldes, Michael let out a small cry as he was delivered and ready for the fight that lay ahead. He was soon whisked away to the neonatal unit where he would take residence for sixty-eight days.
Michael was extremely strong, having been placed initially on CPAP to support his breathing as a precautionary measure, he was breathing by himself within twenty-four hours. My condition however, got progressively worse, being diagnosed with HELLP syndrome, my liver was failing and my blood was not clotting properly.
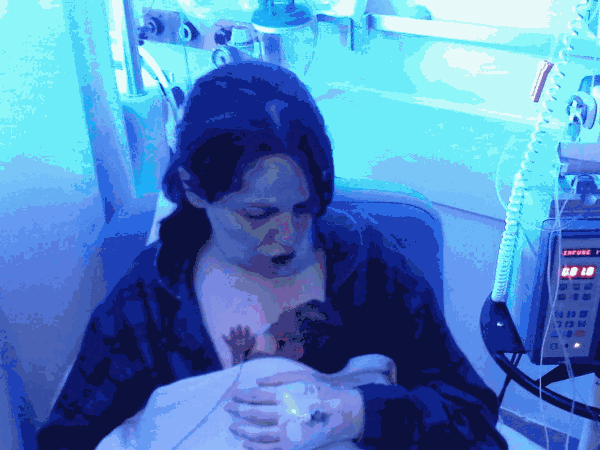
After 24 hours, my condition thankfully stabilised and once able to prove to the midwives that I could get out of bed unaided, I was allowed to go and see Michael. He appeared so fragile, his body covered in many wires and tubes. The nurses carefully took Michael out of the incubator and placed him on my chest for some Kangaroo Care (Skin to skin contact). This was to become a daily routine.
We were warned that a premature baby’s journey in Neonatal was akin to a rollercoaster. During the first week Michael did not tolerate breastmilk and had bile in his stomach. He was required to remain on Total Parental Nutrition (TPN) in which essential food is fed through long lines into a child’s body.
Two weeks after his birth, Michael started to tolerate milk and the Doctors decided that they would remove Michael’s TPN. Something was not right, I could see Michael’s heart rate increasing and his temperature rising on the monitors. Then Michael’s apnoea monitor sounded an alarm, Michael stopped breathing. Emma, the neonatal nurse gently rubbed his back and he started to breathe again. However, Michael stopped breathing again moments later, and having suspected an infection being caused from the TPN line, Emma had already summoned the doctor to assess Michael and address his condition. The Doctors and nurses immediately placed Michael back on to CPAP together with a course of antibiotics in order to fight the infection.
Michael needed to gain weight, this would be a slow process and was always commented upon by doctors during their rounds. Whilst in the incubator, Michael was fed breast milk which had been fortified. His weight soon increased, but once he weighed enough for a cot, his milk was no longer fortified. Unfortunately, whilst in the cot Michael actually lost weight and the hospital assigned a dietician who decided that Michael should consume High Energy SMA.
Michael began increasing in weight with this new milk and was finally discharged from the hospital. Whilst at home, Michael continued with High Energy SMA. Although not a great feeder and being sick often, Michael did put weight on, finally achieving the 25th Centile.
Weaning Michael was difficult and it was during this period we discovered he has quite a strong gag reflux. He would often be sick, even on pureed foods. We had to ensure Michael consumed the correct quantity of food so as to not be sick. Nappy bags soon became a great form of sick bag (and we still carry them around today!), sick bowls would be placed in several locations around the house in case of an emergency.
Michael’s Neonatal consultant closely monitored him, even though he was increasing in weight (albeit very gradually), his height was not increasing. We had always been informed that ‘catch up’ growth would probably happen in the first two years. Michael never caught up with his peers.
At eighteen months old, Michael was no longer taking high energy SMA and instead he was given Fortini supplements. This continued to make Michael sick so we took the decision to stop the supplements. Michael still had support from the dietician until the age of three, but as Michael was at least eating regular meals, the dieticians felt there was nothing further they could do.
When Michael was younger he struggled to fight off infections. When returning home from my first ‘back to work’ day, I noticed that Michael was struggling to breathe and I took him straight to the GP’s who advised to take him straight to A&E if his condition worsened. That night was spent in A&E (a winter was never a winter without a trip to A&E).
At three years of age, the Neonatal Consultant referred Michael to the Endocrinology team at Birmingham Children’s Hospital. We were really hoping that Michael would be discharged from his care (by this time all of the other children who I had met while Michael had been in hospital had been discharged). We never really questioned Michael’s growth before this, we knew he was smaller but there were some possible explanations. Firstly, Michael was born prematurely, secondly, both sets of grandparents are not particularly tall and thirdly we actually believed that he would ‘catch up’, he just needed time.
Michael’s initial Endocrinology appointment was fine, a decision was taken to measure his height accurately for 6 months so that the consultant could monitor him on a height velocity chart to understand his growth rate.
During the next appointment, I felt Michael had grown by less than one inch in 6 months. His health hadn’t been particularly great over the winter period. We discussed the next steps with the consultant and had to take the decision whether to start testing for growth hormone deficiency now or in 6 months time. We made the decision to start the testing right away.
The Stim test concluded that Michael was Growth Hormone Deficient, so a second test was arranged, the arginine test. The results of the second tests resulted in Michael being diagnosed as being Growth Hormone Deficient. On the 31st July 2015 Michael was injected with his first dose of growth hormone. The decision for growth hormone centred on wanting to give Michael an opportunity and if this did not work then we could be satisfied that this avenue had at least been explored.
The first three days were tough with many tears and tantrums. Honesty has been the best policy with Michael by explaining to him why he requires growth hormone. Michael is fantastic with the injections, taking all in his stride without a single complaint.
An MRI scan arranged by the Endocrine team discovered that Michael has a small pituitary gland.
Michael has now managed to be recorded on a growth line, although the 0.4th centile is was a level that he had never reached before. Michael’s health has dramatically improved, no sudden trips to A&E (touch wood) and no need for the frequent use of inhalers.
Michael’s weight is still of concern, having only gained three-and-a-half pounds in one year. We are trying to obtain another referral for a dietician and for a physiotherapist (as his gross motor skills are not at satisfactory levels, Michael struggles to push the pedals of a bike).
Michael is also being checked by a genetics team. A micro array test result was clear, the team are now testing for Russell Silver Syndrome.
Recently Michael had his first operation in which two cysts removed from his mouth. He was incredibly brave and the hospital were fantastic.
Michael is doing extremely well at school and he work very hard. However, he does often get frustrated with his peers because they are too young to realise that every one step of theirs requires two from Michael and all of the various medical appointments or tests that Michael has to endure. Other five year olds do not realise that by calling Michael ‘small’ or ‘a baby’, it affects him and he’ll ask ‘Am I getting bigger? ’.
Michael is now five years old and although he is currently diagnosed as being Growth Hormone Deficient, he is never disheartened and never gives up. A truly courageous and special little boy.
As far as I am aware, the first time the condition showed was in my father, Ramsay. He was born in Birmingham in 1924, the younger of two brothers. His parents and brother (my grandparents and uncle) were of normal size, whereas my father grew to around 4’ 10”. I am not aware that he had any growth treatment as a child or even if anything was available, and unfortunately he is no longer around to ask. The family always said that he had a very bad bout of measles as a child and they thought that this may have been the reason for his growth problems. But as we now know, it is due to a genetic abnormality, or more specifically a gene deletion, which led to the pituitary gland producing insufficient growth hormone. Growing up in the inter-war years would have been difficult for him as ‘he was the first’ with a growth problem, so to speak, so he or his parents would not have known what height he would reach. Also his brother, who was four years older, set various school athletics records at the local grammar school, something that my father could never hope to match when he subsequently went to the same school.
I know his parents took him to the hospital, but at that time the medical advances had not been made so there was nothing that could have been done for him.
A blessing in disguise, if you could call it that, was that he was considered too small to join the army during WWII, although knowing him as I did, I would think that was a cause of severe embarrassment for him as he wanted to serve his country when he was old enough, as his brother was doing.
I don’t believe that he had many girlfriends, but threw himself into music and became a proficient cornet player with the Boys Brigade, which in later years led to the clarinet and saxophone. I’m sure I recall my mother saying once that he hoped to attract a girl through his musical ability, although I think his sense of humour helped as well as him being well read. Incidentally my mother is 5’ 4”.
Pan forward a few years to when I arrived on the scene in 1957. I am the eldest of 3, having 2 sisters born 1959 and 1963. I was told that I was a good weight when I was born and that I grew normally for the first 12 months, but that things started to go wrong soon after that.
That started trips to Birmingham Children’s Hospital; every three months for the next 20 years. It is only now, when writing this, that I realise how much my parents did for me to get me extra height and, from that, what it must have been like for my father as a child, given the determination that I would not have to go through what he did.
By the time I was four my elder sister at two was taller than me. She ended up at 5’ 8” and thin, my younger sister 5’ 7” and thin, me nearly 5’2” and not thin… there’s no justice!
Anyway, back to the story…
Before I started school, I did not know that there was anything wrong, as in those days there were no nurseries to go to for me to compare. I only had three cousins, two of whom were younger, so again no clue of anything wrong.
To be honest I can’t really remember much about my early school days. My mother tells me that with me being small and blonde, all the older girls used to come and pick me up because I was like a doll. Apparently I used to hate it and squirm around to be put down. With the benefit of hindsight, “Girls I forgive you, and I promise I won’t squirm now!”
Other early memories include:
Being useless at anything to do with sport, something that has plagued me throughout my life.
Regular trips to hospital where I was poked & prodded, X-rayed, weighed & measured.
Taking tablets to help me grow (I found out much later that it was an anabolic steroid which was subsequently banned).
Never being able to get clothes to fit properly, particularly trousers, where my mother always had to turn them up (my wife performs that duty now).
Seldom being able to reach anything without steps.
My two younger sisters being a lot taller than me.
My secondary school was an all-boys school, and it was there that I really started to notice the difference in height, as the other lads began to shoot up and of course I didn’t. It was fine with most of the boys in my year but as I got older, the boys from the years below used to make my life a misery, at least when my friends weren’t around. A couple of things that I still remember vividly were:
In Biology we were differentiating the class into various categories and I was the first one to be separated out as being below a certain height.
A new teacher arrived one year and when he came into our classroom for the first time he singled me out as not standing up when he came into the room. Of course I was – which was the cause of much embarrassment on both sides.
Meanwhile, the Children’s Hospital was trying hard to get me a relatively new and rare treatment called Human Growth Hormone. They had run the various tests to see if it would benefit me, which it would, but their request had been turned down a couple of times. Meanwhile I was not getting any younger (I was nearly 14 now). In desperation the doctor in charge of my case gave me the name of a Harley Street doctor who was on the GMC and my parents paid for a private consultation with him.
To cut a long story short, I ended up getting the treatment on the NHS from the age of 14 until I was 21. It was not pleasant having the injections during my latter teenage years especially when I was out with my mates knowing that when I got home I had to have my injection, but I am pleased that I persisted. I would estimate that overall the treatment gave me around 5 inches in height which does not seem much but there is a big difference between 4’ 9” and the 5’ 2” that I am now. Certainly nobody was happier than my father when I eventually overtook him in height (although I never got to be as tall as my mother, and certainly nowhere near my sisters).
Leap forward a number of years, now married and thinking about starting a family. I had already become aware of the CGF due to the problems identified with growth hormone treatment prior to 1985* and was interested in developments within the growth hormone area for the future, in case we decided to have children.
When we decided that the time was right we went to see a genetic specialist to investigate the possibility of passing the condition on to a 3rd generation.
The specialist decided that there was a 50:50 chance, and that it could just as easily happen if the child was a girl, but that my sisters, having not been affected, would not pass the condition on should they have children. Because our case was so rare the specialist took great interest in the family and did blood tests on both my father and I, where it was identified that there was a genetic abnormality causing the pituitary gland to malfunction.
When my wife (Jackie) became pregnant the specialist contacted the hospital where Robert would be born and told them what to look for such as hypoglycaemia. Meanwhile we had attended the CGF national convention and struck up a conversation with a paediatrician sitting next to us at the evening meal who, by complete coincidence, happened to work at the hospital local to where we lived and would subsequently look after Robert.
When he was born it was obvious to the doctors straight away as he was shaking due to hypoglycaemia. He was taken to intensive care and put on a drip to stabilise him. We therefore knew from Day 1 that he had inherited GHD.
The Birmingham Children’s Hospital was brilliant and kept an eye on him. When he was one he had the GH tests, or rather didn’t as the doctor couldn’t get any blood out of him. Despite that the doctor said he had all the hallmarks of GHD and prescribed it anyway.
Robert is now 17 and is in the 6th form at school. He passed 10 GCSEs and achieved the English Baccalaureate. He has just sat AS levels in Maths, Physics, Geography, Business Studies and General Studies.
He is 5’ 7” around 9 ½ stone in weight and is still extremely fit. He no longer runs competitively because of injuries and also because training nights for athletics and rugby coincided. He had to make a choice, so he chose rugby. He played for the 1st XV at school whilst in the lower 6th and will be in the 1st team again for the coming season. He won the award for the most improved player last year, to go with the one from the previous season for the most improved 2nd XV player. In a school 7’s tournament he was the only player in his team to play in every game, because he was the only one fit enough to last out.
He was also drafted in to the senior house hockey team to make up the numbers, (he last played when he was at Junior School) which ended up with the school hockey master asking the rugby master whether he could play hockey for the school team next year…the request was met with a resounding NO!
Outside school he played for Moseley U17 Colts last season as well as Greater Birmingham and will be in the full Colts (U19) squad next season.
In addition to sport, through Scouting he has achieved his Gold & Platinum Chief Scout Awards and his Bronze and Silver Duke of Edinburgh Awards. He is currently in the Lake District for the week doing the Expedition challenge towards the D of E Gold Award which then counts toward his Queen’s Scout.
Other than that, he is learning to drive at the moment and has a girlfriend from the girl’s school next door to his so we will see how that transpires.
*Iatrogenic CJD resulted from the contamination of the very early growth hormone products used before 1985. This contamination of growth hormone is no longer possible due to the completely different method of production.